Structure abstraction#
from rdkit.Chem.Draw import IPythonConsole
IPythonConsole.drawOptions.addAtomIndices = True
To enable using systems and linked structure files in training deep learning models, we’ve implemented a number of useful functions to align, mask, and featurize proteins and ligands.
For this, we convert our PlinderSystem to a Structure object.
from plinder.core import PlinderSystem
plinder_system = PlinderSystem(system_id="4agi__1__1.C__1.W")
system_structure = plinder_system.holo_structure
system_structure
Structure(
(
'id',
'4agi__1__1.C__1.W',
),
(
'protein_path',
/home/runner/.local/share/plinder/2024-06/v2/systems/4agi__1__1.C__1.W/receptor.cif,
),
(
'protein_sequence',
{
'1.C': 'MSTPGAQQVLFRTGIAAVNSTNHLRVYFQDVYGSIRESLYEGSWANGTEKNVIGNAKLGSPVAATSKELKHIRVYTLTEGNTLQEFAYDSGTGWYNGGLGGAKFQVAPYSXIAAVFLAGTDALQLRIYAQKPDNTIQEYMWNGDGWKEGTNLGGALPGTGIGATSFRYTDYNGPSIRIWFQTDDLKLVQRAYDPHKGWYPDLVTIFDRAPPRTAIAATSFGAGNSSIYMRIYFVNSDNTIWQVCWDHGKGYHDKGTITPVIQGSEVAIISWGSFANNGPDLRLYFQNGTYISAVSEWVWNRAHGSQLGRSALPPA',
},
),
(
'ligand_sdfs',
{
'1.W': '/home/runner/.local/share/plinder/2024-06/v2/systems/4agi__1__1.C__1.W/ligand_files/1.W.sdf',
},
),
(
'ligand_smiles',
{
'1.W': 'C[Se][C@@H]1O[C@@H](C)[C@@H](O)[C@@H](O)[C@@H]1O',
},
),
(
'protein_atom_array',
<class 'biotite.structure.AtomArray'> with shape (2443,),
),
(
'ligand_mols',
{
'1.W': (
<rdkit.Chem.rdchem.Mol object at 0x7f3720312420>,
<rdkit.Chem.rdchem.Mol object at 0x7f37202e2880>,
<rdkit.Chem.rdchem.Mol object at 0x7f3720312570>,
(
<class 'numpy.ndarray'> with shape (1, 12),
<class 'numpy.ndarray'> with shape (1, 12),
),
),
},
),
(
'add_ligand_hydrogens',
False,
),
(
'skip_3d_confgen',
False,
),
(
'structure_type',
'holo',
),
)
We list all fields and their
FieldInfoto show which ones are required.id,protein_pathandprotein_sequenceare required. Everything else is optionally. Particularly worth mentioning is the decision to makelist_ligand_sdf_and_input_smilesoptional; this is because ligand will not be availbale in apo and predicted structures.Out of these field
ligand_molsandprotein_atom_arrayis computed within the object if set to default.ligand_molsreturns a chain-mapped dictionary of of the form:{ "<instance_id>.<chain_id>": ( RDKit 2D mol from template SMILES of type `Chem.Mol`, RDKit mol from template SMILES with random 3D conformer of type `Chem.Mol`, RDKit mol of solved (holo) ligand structure of type `Chem.Mol`, paired stacked arrays (template vs holo) mapping atom order by index of type `tuple[NDArray.int_, NDArray.int_]` ) }While
protein_atom_arrayreturns biotite AtomArray of the receptor protein structure.add_ligand_hydrogensspecifies whether to adds hydrogens to ligandstructure_type: could be"holo","apo"or"pred"
system_structure.model_fields
{'id': FieldInfo(annotation=str, required=True),
'protein_path': FieldInfo(annotation=Path, required=True),
'protein_sequence': FieldInfo(annotation=Union[dict[str, str], NoneType], required=False, default=None),
'ligand_sdfs': FieldInfo(annotation=Union[dict[str, str], NoneType], required=False, default=None),
'ligand_smiles': FieldInfo(annotation=Union[dict[str, str], NoneType], required=False, default=None),
'protein_atom_array': FieldInfo(annotation=Union[AtomArray, NoneType], required=False, default=None),
'ligand_mols': FieldInfo(annotation=Union[dict[str, tuple[Mol, Mol, Mol, tuple[ndarray[Any, dtype[+_ScalarType_co]], ndarray[Any, dtype[+_ScalarType_co]]]]], NoneType], required=False, default=None),
'add_ligand_hydrogens': FieldInfo(annotation=bool, required=False, default=False),
'skip_3d_confgen': FieldInfo(annotation=bool, required=False, default=False),
'structure_type': FieldInfo(annotation=str, required=False, default='holo')}
Ligand#
for name in system_structure.get_properties():
if "ligand" in name:
print(name)
input_ligand_conformer_atom_array
input_ligand_conformers
input_ligand_conformers_coords
input_ligand_templates
ligand_atom_array
ligand_chain_ordered
ligand_template2resolved_atom_order_stacks
resolved_ligand_mols
resolved_ligand_mols_coords
The ligands are provided using dictionaries.
These dictionaries contain information for each ligand:
input_ligand_templates: 2D RDKit mols generated from the RDKit canonical SMILESinput_ligand_conformers: 3D (random) conformers generated for each input molinput_ligand_conformers_coords: positional coordintates for 3D conformersresolved_ligand_mols: RDKit mols of solved (holo) ligand structuresresolved_ligand_mols_coords: positional coordintates for holo ligand structuresligand_template2resolved_atom_order_stacks: paired stacked arrays (template vs holo) mapping atom order by indexligand_chain_ordered: ordered list of all ligands by their keys
Ligand atom id mapping mapping#
Unlike the protein sequence - there is no canonical order to ligand atoms in the molecule. It can be further complicated by automorphisms present in the structure due to symmetry, i.e. there is more than one match that is possible between the structures.
This is important when calculating ligand structure loss, as the most optimal atom order can change between the different inference results. Typically, it is accepted to take the atom ordering resulting in the best objective score and use that for the loss calculation.
Occasionally futher ambiguity arises to to part of the ligand structure being unresolved in the holo structure - this can lead to multiple available matches. We use RascalMCES algorithm from RDKit to provide all the possible matches between the atom order in the input structure (from SMILES) to the resolved holo structure.
This is provided as stacks of atom order arrays that reorder the template and holo indices to provide matches. Each stack is a unique order transformation and should be iterated.
system_structure.input_ligand_templates[system_structure.ligand_chain_ordered[0]]
system_structure.input_ligand_conformers[system_structure.ligand_chain_ordered[0]]
system_structure.resolved_ligand_mols[system_structure.ligand_chain_ordered[0]]
Ligand conformer coordinates#
As you can tell, the input 2D and 3D conformer indices match, but the resolved ligand is different. Thus to perform a correct comparison for their coordinates one should use atom order stacks.
(
input_atom_order_stack,
holo_atom_order_stack,
) = system_structure.ligand_template2resolved_atom_order_stacks[
system_structure.ligand_chain_ordered[0]
]
input_atom_order_stack, holo_atom_order_stack
(array([[ 2, 10, 8, 6, 4, 5, 11, 9, 7, 3, 0, 1]]),
array([[ 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11]]))
system_structure.input_ligand_conformers_coords[
system_structure.ligand_chain_ordered[0]
][input_atom_order_stack]
array([[[-0.40725066, -1.47748498, 0.185955 ],
[ 1.0643663 , -1.31706023, 0.33981289],
[ 1.51382048, -0.06269305, 1.01334722],
[ 0.43775803, 1.01633012, 1.02753193],
[-0.32555831, 0.7522505 , -0.26447634],
[-1.0585462 , 1.94667957, -0.76103278],
[ 1.730437 , -1.48907463, -0.8864381 ],
[ 2.0006517 , -0.2496771 , 2.29537409],
[-0.4092932 , 0.72044372, 2.10546384],
[-1.13277182, -0.33267599, 0.01577224],
[-2.16322047, -1.83693988, -2.49576751],
[-0.84673963, -2.66539344, -1.30355921]]])
system_structure.resolved_ligand_mols_coords[system_structure.ligand_chain_ordered[0]][
holo_atom_order_stack
]
array([[[48.954, 28.186, 62.068],
[48.681, 29.643, 61.777],
[48.556, 29.933, 60.282],
[47.499, 28.988, 59.699],
[47.948, 27.56 , 59.939],
[46.992, 26.503, 59.374],
[49.72 , 30.41 , 62.383],
[48.255, 31.352, 60.148],
[46.262, 29.193, 60.366],
[48.031, 27.383, 61.339],
[50.647, 25.895, 62.426],
[50.853, 27.716, 61.696]]])
More complicated examples#
Symmetry in ligand (automorphism) - two ways of pairwise mapping the atom order
structure_with_symmetry_in_ligand = PlinderSystem(
system_id="4v2y__1__1.A__1.E"
).holo_structure
structure_with_symmetry_in_ligand.input_ligand_templates[
structure_with_symmetry_in_ligand.ligand_chain_ordered[0]
]
structure_with_symmetry_in_ligand.resolved_ligand_mols[
structure_with_symmetry_in_ligand.ligand_chain_ordered[0]
]
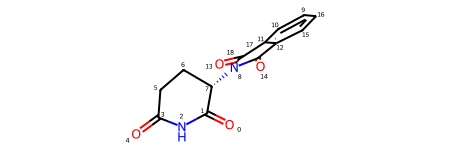
structure_with_symmetry_in_ligand.ligand_template2resolved_atom_order_stacks[
structure_with_symmetry_in_ligand.ligand_chain_ordered[0]
]
(array([[17, 16, 18, 1, 0, 2, 3, 4, 5, 10, 9, 8, 13, 7, 15, 12,
11, 14, 6],
[17, 16, 18, 1, 0, 2, 3, 4, 5, 11, 12, 13, 8, 15, 7, 9,
10, 6, 14]]),
array([[ 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,
16, 17, 18],
[ 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,
16, 17, 18]]))
Symmetry arises due to ligand being partially resolved - there are three template pieces that can be mapped to the resolved ground truth.
structure_with_partly_resolved_ligand = PlinderSystem(
system_id="1ngx__1__1.A_1.B__1.E"
).holo_structure
structure_with_partly_resolved_ligand.input_ligand_templates[
structure_with_partly_resolved_ligand.ligand_chain_ordered[0]
]
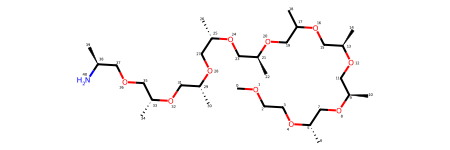
structure_with_partly_resolved_ligand.resolved_ligand_mols[
structure_with_partly_resolved_ligand.ligand_chain_ordered[0]
]
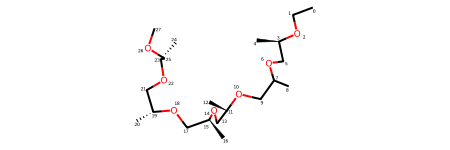
structure_with_partly_resolved_ligand.ligand_template2resolved_atom_order_stacks[
structure_with_partly_resolved_ligand.ligand_chain_ordered[0]
]
(array([[ 9, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25,
26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37],
[ 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17,
18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29],
[ 5, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,
22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33]]),
array([[ 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,
16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27],
[ 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,
16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27],
[ 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,
16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27]]))
Protein#
for name in system_structure.get_properties():
if "protein" in name:
print(name)
protein_backbone_mask
protein_calpha_coords
protein_calpha_mask
protein_chain_ordered
protein_chains
protein_coords
protein_n_atoms
protein_sequence_from_structure
protein_structure_b_factor
protein_structure_tokenized_sequence
protein_unique_atom_names
protein_unique_residue_ids
protein_unique_residue_names
Others properties#
This includes:
num_ligands: Number of ligand chainssmiles: Ligand smiles dictionarynum_proteins: Number of protein chains
Masking#
The properties protein_backbone_mask and protein_calpha_mask are boolean masks that can be used to select backbone or calpha atoms from biotite AtomArray. The indices of True corresponds to backbone or calpha indices.
print(
"Total number of atoms:",
len(system_structure.protein_atom_array),
)
print("Number of backbone atoms:", system_structure.protein_backbone_mask.sum())
print(
"Number of calpha atoms:",
system_structure.protein_calpha_mask.sum(),
)
Total number of atoms: 2443
Number of backbone atoms: 942
Number of calpha atoms: 314
calpha_atom_array = system_structure.protein_atom_array[
system_structure.protein_calpha_mask
]
calpha_atom_array.coord.shape
(314, 3)
You can also filter by arbitrary properties of the AtomArray using the filter method. This returns a new Structure object.
calpha_structure = system_structure.filter(
property="atom_name",
mask="CA",
)
calpha_structure.protein_atom_array.coord.shape
(314, 3)
Get protein chain ordered#
This gives a list of protein chains ordered by how they are in the structure
system_structure.protein_chain_ordered
['1.C']
Get protein chains for all atoms#
The list of chain IDs in the structure. Order of how they appear not kept.
system_structure.protein_chains
['1.C']
Get protein coordinates#
This property gets the 3D positions of each of the atoms in protein molecules
system_structure.protein_coords
[array([[48.761, 52.015, 58.719],
[48.178, 50.968, 57.827],
[49.186, 50.593, 56.712],
...,
[50.066, 42.485, 54.198],
[47.368, 43.433, 55.544],
[50.022, 44.674, 54.248]], dtype=float32)]
Get number of atoms of protein molecule#
system_structure.protein_n_atoms
2443
Get protein structure atom names#
Returns all atoms names the same way they appear in the structure
system_structure.protein_unique_atom_names
['C',
'CA',
'CB',
'CD',
'CD1',
'CD2',
'CE',
'CE1',
'CE2',
'CE3',
'CG',
'CG1',
'CG2',
'CH2',
'CM',
'CZ',
'CZ2',
'CZ3',
'N',
'ND1',
'ND2',
'NE',
'NE1',
'NE2',
'NH1',
'NH2',
'NZ',
'O',
'OD1',
'OD2',
'OE1',
'OE2',
'OG',
'OG1',
'OH',
'OXT',
'SD',
'SE',
'SG']
Get protein residue names#
system_structure.protein_unique_residue_names
['ALA',
'ARG',
'ASN',
'ASP',
'CSD',
'CSK',
'GLN',
'GLU',
'GLY',
'HIS',
'ILE',
'LEU',
'LYS',
'MET',
'PHE',
'PRO',
'SER',
'THR',
'TRP',
'TYR',
'VAL']
Get protein residues number#
Residue number as they appear in structure
system_structure.protein_unique_residue_ids
[2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28,
29,
30,
31,
32,
33,
34,
35,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49,
50,
51,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72,
73,
74,
75,
76,
77,
78,
79,
80,
81,
82,
83,
84,
85,
86,
87,
88,
89,
90,
91,
92,
93,
94,
95,
96,
97,
98,
99,
100,
101,
102,
103,
104,
105,
106,
107,
108,
109,
110,
111,
112,
113,
114,
115,
116,
117,
118,
119,
120,
121,
122,
123,
124,
125,
126,
127,
128,
129,
130,
131,
132,
133,
134,
135,
136,
137,
138,
139,
140,
141,
142,
143,
144,
145,
146,
147,
148,
149,
150,
151,
152,
153,
154,
155,
156,
157,
158,
159,
160,
161,
162,
163,
164,
165,
166,
167,
168,
169,
170,
171,
172,
173,
174,
175,
176,
177,
178,
179,
180,
181,
182,
183,
184,
185,
186,
187,
188,
189,
190,
191,
192,
193,
194,
195,
196,
197,
198,
199,
200,
201,
202,
203,
204,
205,
206,
207,
208,
209,
210,
211,
212,
213,
214,
215,
216,
217,
218,
219,
220,
221,
222,
223,
224,
225,
226,
227,
228,
229,
230,
231,
232,
233,
234,
235,
236,
237,
238,
239,
240,
241,
242,
243,
244,
245,
246,
247,
248,
249,
250,
251,
252,
253,
254,
255,
256,
257,
258,
259,
260,
261,
262,
263,
264,
265,
266,
267,
268,
269,
270,
271,
272,
273,
274,
275,
276,
277,
278,
279,
280,
281,
282,
283,
284,
285,
286,
287,
288,
289,
290,
291,
292,
293,
294,
295,
296,
297,
298,
299,
300,
301,
302,
303,
304,
305,
306,
307,
308,
309,
310,
311,
312,
313,
314,
315]
Get sequence from protein structure#
system_structure.protein_sequence_from_structure
'STPGAQQVLFRTGIAAVNSTNHLRVYFQDVYGSIRESLYEGSWANGTEKNVIGNAKLGSPVAATSKELKHIRVYTLTEGNTLQEFAYDSGTGWYNGGLGGAKFQVAPYSXIAAVFLAGTDALQLRIYAQKPDNTIQEYMWNGDGWKEGTNLGGALPGTGIGATSFRYTDYNGPSIRIWFQTDDLKLVQRAYDPHKGWYPDLVTIFDRAPPRTAIAATSFGAGNSSIYMRIYFVNSDNTIWQVCWDHGKGYHDKGTITPVIQGSEVAIISWGSFANNGPDLRLYFQNGTYISAVSEWVWNRAHGSQLGRSALPPA'
Note that this is different from the canonical SEQRES sequence due to unresolved terminal residues:
system_structure.protein_sequence
{'1.C': 'MSTPGAQQVLFRTGIAAVNSTNHLRVYFQDVYGSIRESLYEGSWANGTEKNVIGNAKLGSPVAATSKELKHIRVYTLTEGNTLQEFAYDSGTGWYNGGLGGAKFQVAPYSXIAAVFLAGTDALQLRIYAQKPDNTIQEYMWNGDGWKEGTNLGGALPGTGIGATSFRYTDYNGPSIRIWFQTDDLKLVQRAYDPHKGWYPDLVTIFDRAPPRTAIAATSFGAGNSSIYMRIYFVNSDNTIWQVCWDHGKGYHDKGTITPVIQGSEVAIISWGSFANNGPDLRLYFQNGTYISAVSEWVWNRAHGSQLGRSALPPA'}
Get tokenized sequence#
Get tensor of sequence converted to integer-based amino acid tokens
system_structure.protein_structure_tokenized_sequence
tensor([15, 16, 14, 7, 0, 5, 5, 19, 10, 13, 1, 16, 7, 9, 0, 0, 19, 2,
15, 16, 2, 8, 10, 1, 19, 18, 13, 5, 3, 19, 18, 7, 15, 9, 1, 6,
15, 10, 18, 6, 7, 15, 17, 0, 2, 7, 16, 6, 11, 2, 19, 9, 7, 2,
0, 11, 10, 7, 15, 14, 19, 0, 0, 16, 15, 11, 6, 10, 11, 8, 9, 1,
19, 18, 16, 10, 16, 6, 7, 2, 16, 10, 5, 6, 13, 0, 18, 3, 15, 7,
16, 7, 17, 18, 2, 7, 7, 10, 7, 7, 0, 11, 13, 5, 19, 0, 14, 18,
15, 20, 9, 0, 0, 19, 13, 10, 0, 7, 16, 3, 0, 10, 5, 10, 1, 9,
18, 0, 5, 11, 14, 3, 2, 16, 9, 5, 6, 18, 12, 17, 2, 7, 3, 7,
17, 11, 6, 7, 16, 2, 10, 7, 7, 0, 10, 14, 7, 16, 7, 9, 7, 0,
16, 15, 13, 1, 18, 16, 3, 18, 2, 7, 14, 15, 9, 1, 9, 17, 13, 5,
16, 3, 3, 10, 11, 10, 19, 5, 1, 0, 18, 3, 14, 8, 11, 7, 17, 18,
14, 3, 10, 19, 16, 9, 13, 3, 1, 0, 14, 14, 1, 16, 0, 9, 0, 0,
16, 15, 13, 7, 0, 7, 2, 15, 15, 9, 18, 12, 1, 9, 18, 13, 19, 2,
15, 3, 2, 16, 9, 17, 5, 19, 4, 17, 3, 8, 7, 11, 7, 18, 8, 3,
11, 7, 16, 9, 16, 14, 19, 9, 5, 7, 15, 6, 19, 0, 9, 9, 15, 17,
7, 15, 13, 0, 2, 2, 7, 14, 3, 10, 1, 10, 18, 13, 5, 2, 7, 16,
18, 9, 15, 0, 19, 15, 6, 17, 19, 17, 2, 1, 0, 8, 7, 15, 5, 10,
7, 1, 15, 0, 10, 14, 14, 0])
Linked protein input structures#
For realistic inference scenarios we need to initialize our protein structures using a linked structure (introduced above). In most cases, these will not be a perfect match to the holo structure - the number of residues, residue numbering, and sometime the sequnce can be different. It’s important to be able to match these structures to ensure that we can map between them.
In the example below, we will take a holo structure and it’s linked predicted (pred) form with different number of residues and match and crop the resulting structures to figure out the correspondence between their residues. For this we use the align_common_sequence function of the holo Structure object, which aligns two structures based on their shared sequences. It has the following parameters:
other: Structure
The other structure to align to
copy: bool
Whether to make a copy or edit in-place
remove_differing_atoms: bool
Whether to remove differing atoms between the two structure
renumber_residues: bool [False]
If True, renumber residues in the two structures to match and starting from 1.
If False, sets the resulting residue indices to the one from the aligned sequence
remove_differing_annotations: bool [False]
Whether to remove differing annotations, like b-factor, etc
In this example, we will match, make copies and crop the structures.
Note
To use this function the proteins to be aligned must have the same chain ids. So, we first set the chain id of the predicted structure to that of the holo structure. :::
plinder_system = PlinderSystem(system_id="4cj6__1__1.A__1.B")
holo = plinder_system.holo_structure
predicted = plinder_system.alternate_structures["P12271_A"]
predicted.set_chain(holo.protein_chain_ordered[0])
holo_cropped, predicted_cropped = holo.align_common_sequence(predicted)
predicted_cropped_superposed, raw_rmsd, refined_rmsd = predicted_cropped.superimpose(
holo_cropped
)