Pipeline#
We outline conceptually the steps of the end-to-end pipeline in the following sections. We briefly describe some of the abstractions that are used to orchestrate the entire pipeline, but they are to be considered an implementation detail because they rely on our choice of orchestration framework for job execution.
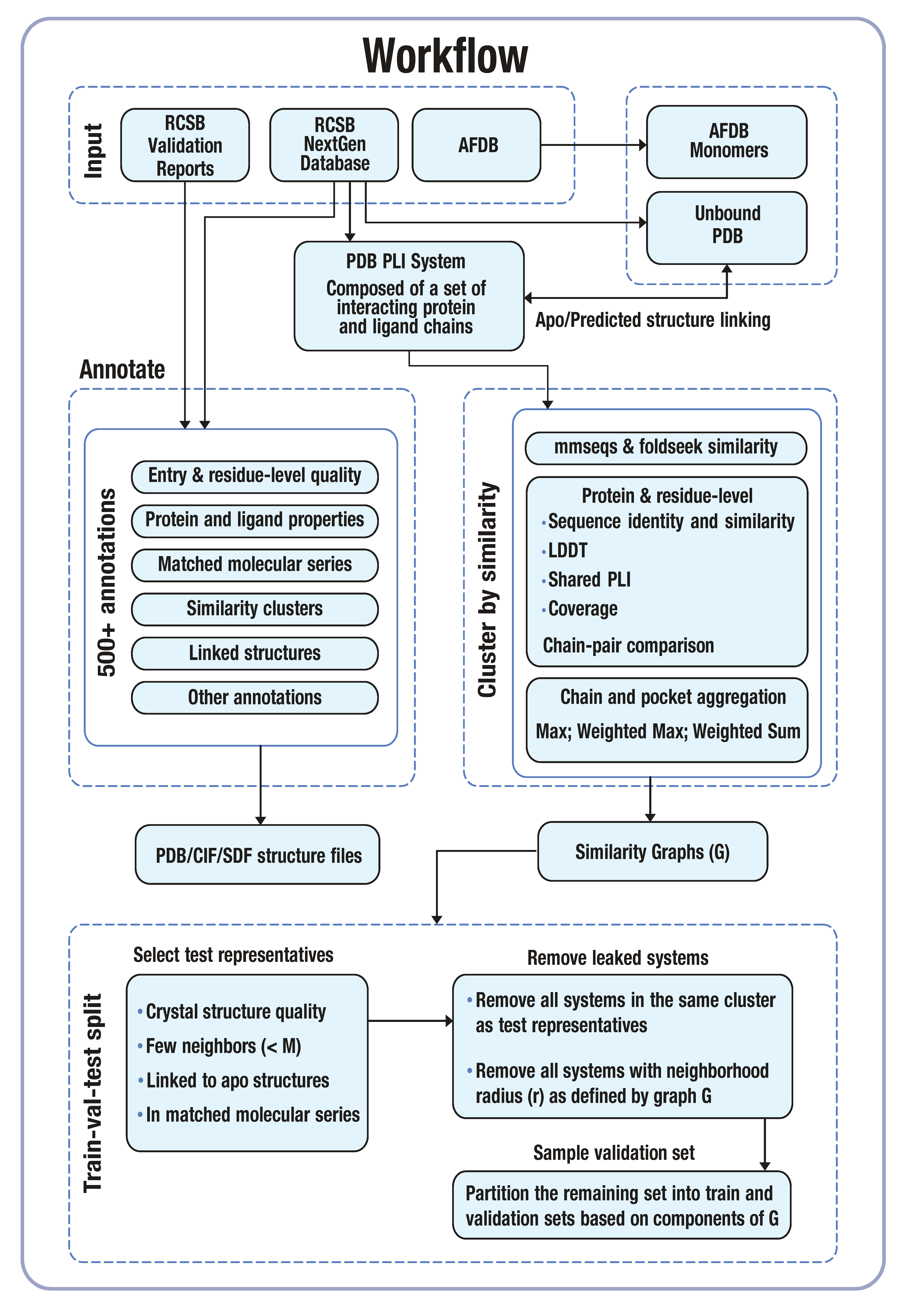
Ingestion#
The code to obtain the raw data sources used in plinder are housed
in the plinder.data.pipeline.io package and are invoked in our
end-to-end pipeline through task wrappers in plinder.data.pipeline.tasks.
tasks.download_rcsb_files: uses the RCSB rsync API to download the majority of the raw dataThis is a distributed task that is called in parallel for chunks of two character codes
It syncs both the next-gen
cif.gzand validationxml.gzfiles for all entriesSide effects include writing the following files:
ingest/{two_char_code}/{full_pdb_id}/{full_pdb_id}-enrich.cif.gzreports/{two_char_code}/{pdb_id}/{pdb_id}_validation.xml.gz
tasks.download_alternative_datasets: download all the alternative datasets used to enrichplinderThis is a task that is called once but reaches out to numerous external REST APIs
This could be threaded and arguably the alphafold sync could be its own task
Side effects include writing the following files:
dbs/alphafold/AF-{uniprod_id}-F1-model_v4.cifdbs/cofactors/cofactors.jsondbs/components/components.parquetdbs/ecod/ecod.parquetdbs/kinase/kinase_information.parquetdbs/kinase/kinase_ligand_ccd_codes.parquetdbs/kinase/kinase_uniprotac.parquetdbs/kinase/kinase_structures.parquetdbs/panther/panther_{i}.parquetdbs/seqres/pdb_seqres.txt.gz
Database creation#
Once the raw data is downloaded, we need to create the foldseek and mmseqs
databases to be used as a basis for the similarity datasets.
tasks.make_dbs: creates thefoldseekandmmseqsdatabasesThis is a task that is called once
It uses the
cif.gzdata to create thefoldseekdatabaseIt uses the
pdb_seqres.txt.gzdata to create themmseqsdatabase (obtained indownload_alternative_datasets)
Annotation generation#
Once the raw data is downloaded, we can start generating the annotation data. Technically, this could run in parallel with the database creation, but it this task is already heavily distributed and it would add complexity to the DAG.
tasks.make_entries: creates theraw_entriesdataThis is a distributed task that is called in parallel for chunks of PDB IDs
It uses the
cif.gzandxml.gzdata inEntry.from_cif_fileIt additionally uses the following alternative datasets:
ECODPantherKinaseCofactorsComponents
Side effects include writing the following files:
raw_entries/{two_char_code}/{pdb_id}.jsonraw_entries/{two_char_code}/{system_id}/**
Structure quality checks#
After raw annotation generation, we run a series of quality checks on the generated data and do some consolidation and organization of the generated data.
tasks.structure_qc: runs the structure quality checksThis is a distributed task that is called in parallel for chunks of two character codes
It reads in the JSON files in
raw_entriesSide effects include writing the following files:
qc/index/{two_char_code}.parquet- consolidated annotationsqc/logs/{two_char_code}_qc_fails.csv- list of entries that failed QCentries/{two_char_code}.zip- zipped JSON entries
Structure archives#
The amount of structural data generated in make_entries is large and consolidated
separately in its own task.
tasks.make_system_archives: creates the structure archivesThis is a distributed task that is called in parallel for chunks of two character codes
It consolidates the structure files into zip archives in the same layout as for
entriesThe inner structure of each structure zip is grouped by system ID
Side effects include writing the following files:
archives/{two_char_code}.zip- zipped structure files
Ligand Similarity#
Once the plinder systems have been generated by make_entries, we can enumerate
the small molecule ligands in the dataset.
tasks.make_ligands: creates theligandsdataThis is a distributed task that is called in parallel for chunks of PDB IDs
It filters out ligands that are acceptable for use in ligand similarity
It uses the JSON files from
raw_entriesSide effects include writing the following files:
ligands/{chunk_hash}.parquet
tasks.compute_ligand_fingerprints: computes the ligand fingerprintsThis is a task that is called once
It uses the
ligandsdataSide effects include writing the following files:
fingerprints/ligands_per_inchikey.parquetfingerprints/ligands_per_inchikey_ecfp4.npyfingerprints/ligands_per_system.parquet
tasks.make_ligand_scores: creates theligand_scoresdataThis is a distributed task that is called in parallel for chunks of ligand IDs
It uses the
fingerprintsdataSide effects include writing the following files:
ligand_scores/{fragment}.parquet
Sub-databases#
Once the plinder systems have been generated, we are able to split the foldseek
and mmseqs databases into sub-databases containing holo and apo systems. We additionally use the alphafold linked_structures
to create the pred sub-database.
tasks.make_sub_dbs: creates theholoandaposub-databasesThis is a task that is called once
It uses the
foldseekandmmseqsdatabasesSide effects include writing the following files:
dbs/subdbs/holo_foldseek/**dbs/subdbs/apo_foldseek/**dbs/subdbs/pred_foldseek/**dbs/subdbs/holo_mmseqs/**dbs/subdbs/apo_mmseqs/**dbs/subdbs/pred_mmseqs/**
Protein similarity#
With the holo and apo sub-databases created, we can run
protein similarity scoring for all plinder systems.
tasks.run_batch_searches: runs thefoldseekandmmseqssearches for large batchesThis is a distributed task that is called in parallel for large chunks of PDB IDs
It uses the JSON files from
raw_entriesand theholoandaposub-databasesSide effects include writing the following files:
foldseekandmmseqssearch results
tasks.make_batch_scores: creates the protein similarity scoresThis is a distributed task that is called in parallel for smaller chunks of PDB IDs
It uses the JSON files from
raw_entriesand thefoldseekandmmseqssearch resultsSide effects include writing the following files:
scores/search_db=holo/*scores/search_db=apo/*scores/search_db=pred/*
MMP and MMS#
tasks.make_mmp_index: creates themmpdatasetThis is a task that is called once
It additionally consolidates the
indexdataset created instructure_qcinto a single parquet fileSide effects include writing the following files:
mmp/plinder_mmp_series.parquetmmp/plinder_mms.csv.gz
Clustering#
Once the protein similarity scores are generated, we run component and community clustering.
tasks.make_components_and_communities: creates thecomponentsandcommunitiesclusters for given metrics at given thresholdsThis is a distributed task that is called in parallel for individual tuples of metric and threshold
It uses the protein similarity scores and the annotation index
Side effects include writing the following files:
clusters/**
Splits#
Armed with the clusters from the previous step, we can now split the plinder systems into train, test and val.
Leakage#
With splits in hand, we perform an exhaustive evaluation of the generated splits to quantify the quality of the splits through leakage metrics.
Technical details#
Schemas#
The scores protein similarity dataset is a collection of
parquet files with the following schema:
>>> from plinder.data.schemas import PROTEIN_SIMILARITY_SCHEMA
>>> PROTEIN_SIMILARITY_SCHEMA
query_system: string
target_system: string
protein_mapping: string
mapping: string
protein_mapper: dictionary<values=string, indices=int8, ordered=0>
source: dictionary<values=string, indices=int8, ordered=1>
metric: dictionary<values=string, indices=int8, ordered=1>
similarity: int8
The ligand_scores ligand similarity dataset is a collection of
parquet files with the following schema:
>>> from plinder.data.schemas import TANIMOTO_SCORE_SCHEMA
>>> TANIMOTO_SCORE_SCHEMA
query_ligand_id: int32
target_ligand_id: int32
tanimoto_similarity_max: int8
The clusters clustering dataset is a collection of
parquet files with the following schema:
>>> from plinder.data.schemas import CLUSTER_DATASET_SCHEMA
>>> CLUSTER_DATASET_SCHEMA
metric: string
directed: bool
threshold: int8
system_id: string
component: string
The splits split datasets are independent
parquet files with the following schema:
>>> from plinder.data.schemas import SPLIT_DATASET_SCHEMA
>>> SPLIT_DATASET_SCHEMA
system_id: string
split: string
cluster: string
cluster_for_val_split: string
The linked_structures datasets are independent
parquet files with the following schema:
>>> from plinder.data.schemas import STRUCTURE_LINK_SCHEMA
>>> STRUCTURE_LINK_SCHEMA
query_system: string
target_system: string
protein_qcov_weighted_sum: float
protein_fident_weighted_sum: float
pocket_fident: float
target_id: string
sort_score: float